
![]()
What is conos? Conos is an R package to wire together large collections of single-cell RNA-seq datasets, which allows for both the identification of recurrent cell clusters and the propagation of information between datasets in multi-sample or atlas-scale collections. It focuses on the uniform mapping of homologous cell types across heterogeneous sample collections. For instance, users could investigate a collection of dozens of peripheral blood samples from cancer patients combined with dozens of controls, which perhaps includes samples of a related tissue such as lymph nodes.
How does it work? 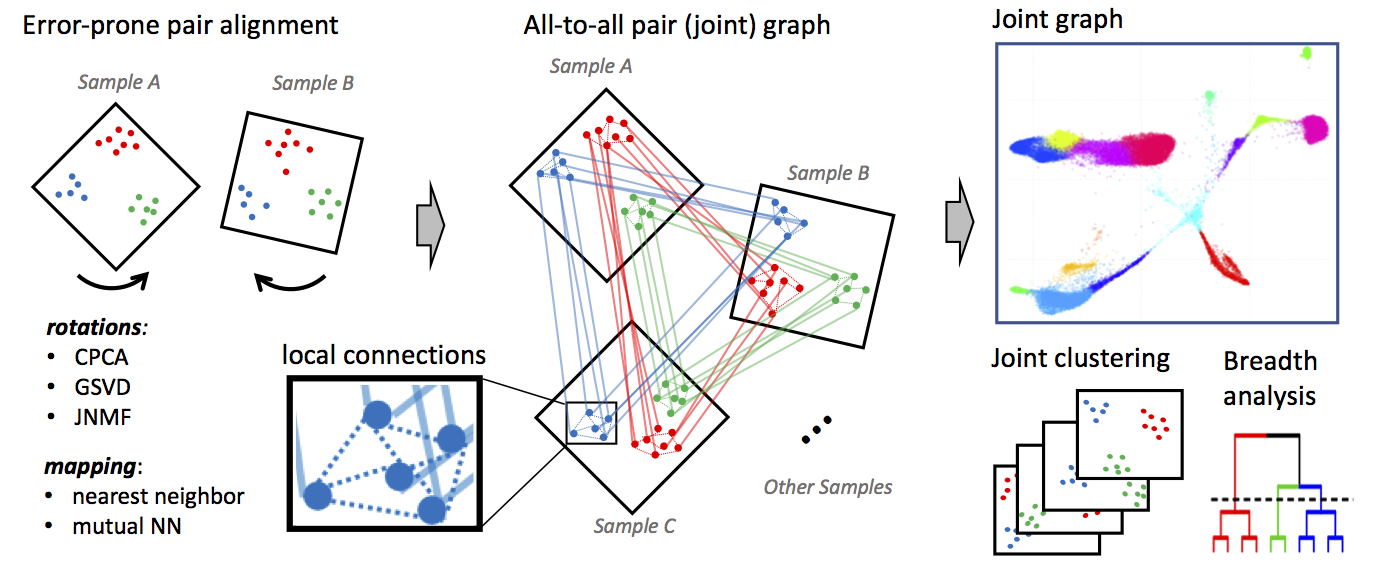 Conos applies one of many error-prone methods to align
each pair of samples in a collection, establishing weighted inter-sample
cell-to-cell links. The resulting joint graph can then be analyzed to
identify subpopulations across different samples. Cells of the same type
will tend to map to each other across many such pairwise comparisons,
forming cliques that can be recognized as clusters (graph
communities).
Conos applies one of many error-prone methods to align
each pair of samples in a collection, establishing weighted inter-sample
cell-to-cell links. The resulting joint graph can then be analyzed to
identify subpopulations across different samples. Cells of the same type
will tend to map to each other across many such pairwise comparisons,
forming cliques that can be recognized as clusters (graph
communities).
Conos processing can be divided into three phases:
pagoda2 or Seurat. Specifically, Conos relies
on these methods to perform cell filtering, library size normalization,
identification of overdispersed genes and, in the case of pagoda2,
variance normalization. (Conos is robust to variations in the
normalization procedures, but it is recommended that all of the datasets
be processed uniformly.)What does it produce? In essence, conos will
take a large, potentially heterogeneous panel of samples and will
produce clustering grouping similar cell subpopulations together in a
way that will be robust to inter-sample variation:
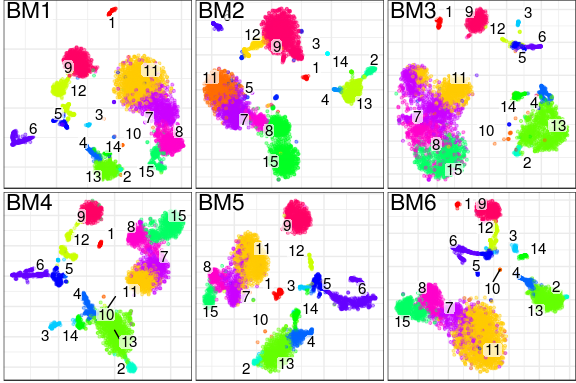
What are the advantages over existing alignment methods? Conos is robust to heterogeneity of samples within a collection, as well as noise. The ability to resolve finer subpopulation structure improves as the size of the panel increases.
Given a list of individual processed samples (pl), conos
processing can be as simple as this:
# Construct Conos object, where pl is a list of pagoda2 objects
con <- Conos$new(pl)
# Build joint graph
con$buildGraph()
# Find communities
con$findCommunities()
# Generate embedding
con$embedGraph()
# Plot joint graph
con$plotGraph()
# Plot panel with joint clustering results
con$plotPanel()To see more documentation on the class Conos, run
?Conos.
Please see the following tutorials for detailed examples of how to use conos:
Note that for integration with Scanpy, users need to save conos files to disk from an R session, and then load these files into Python.
Save conos for Scanpy: * HTML version * Markdown version
Load conos files into Scanpy: * Jupyter Notebook
First of all, in order to obtain an RNA velocity plot from a
Conos object you have to use the dropEst pipeline to
align and annotate your single-cell RNA-seq measurements. You can see this
tutorial and this
shell script to see how it can be done. In this example we
specifically assume that when running dropEst you have used the
-V option to get estimates of unspliced/spliced counts
from the dropEst directly. Secondly, you need the velocyto.R package for the actual
velocity estimation and visualisation.
After running dropEst you should have 2 files for each of the
samples: - sample.rds (matrix of counts) -
sample.matrices.rds (3 matrices of exons, introns and
spanning reads)
The .matrices.rds files are the velocity files. Load
them into R in a list (same order as you give to conos). Load,
preprocess and integrate with conos the count matrices
(.rds) as you normally would. Before running the velocity,
you must at least create an embedding and run the leiden clustering.
Finally, you can estimate the velocity as follows:
### Assuming con is your Conos object and cms.list is the list of your velocity files ###
library(velocyto.R)
# Preprocess the velocity files to match the Conos object
vi <- velocityInfoConos(cms.list = cms.list, con = con,
n.odgenes = 2e3, verbose = TRUE)
# Estimate RNA velocity
vel.info <- vi %$%
gene.relative.velocity.estimates(emat, nmat, cell.dist = cell.dist,
deltaT = 1, kCells = 25, fit.quantile = 0.05, n.cores = 4)
# Visualise the velocity on your Conos embedding
# Takes a very long time!
# Assign to a variable to speed up subsequent recalculations
cc.velo <- show.velocity.on.embedding.cor(vi$emb, vel.info, n = 200, scale = 'sqrt',
cell.colors = ac(vi$cell.colors, alpha = 0.5),
cex = 0.8, grid.n = 50, cell.border.alpha = 0,
arrow.scale = 3, arrow.lwd = 0.6, n.cores = 4,
xlab = "UMAP1", ylab = "UMAP2")
# Use cc=cc.velo$cc when running again (skips the most time consuming delta projections step)
show.velocity.on.embedding.cor(vi$emb, vel.info, cc = cc.velo$cc, n = 200, scale = 'sqrt',
cell.colors = ac(vi$cell.colors, alpha = 0.5),
cex = 0.8, arrow.scale = 15, show.grid.flow = TRUE,
min.grid.cell.mass = 0.5, grid.n = 40, arrow.lwd = 2,
do.par = F, cell.border.alpha = 0.1, n.cores = 4,
xlab = "UMAP1", ylab = "UMAP2")To install the stable version from CRAN, use:
install.packages('conos')To install the latest version of conos, use:
install.packages('devtools')
devtools::install_github('kharchenkolab/conos')The dependencies are inherited from pagoda2. Note that this package also has the dependency igraph, which requires various libraries to install correctly. Please see the installation instructions at that page for more details, along with the github README here.
To install system dependencies using apt-get, use the
following:
sudo apt-get update
sudo apt-get -y install libcurl4-openssl-dev libssl-dev libxml2-dev libgmp-dev libglpk-devFor Red Hat distributions using yum, use the following
command:
sudo yum update
sudo yum install openssl-devel libcurl-devel libxml2-devel gmp-devel glpk-develUsing the Mac OS package manager Homebrew, try the following command:
brew update
brew install openssl curl-openssl libxml2 glpk gmp(You may need to run brew uninstall curl in order for
brew install curl-openssl to be successful.)
As of version 1.3.1, conos should successfully install
on Mac OS. However, if there are issues, please refer to the following
wiki page for further instructions on installing conos with
Mac OS: Installing
conos for Mac OS
If your system configuration is making it difficult to install
conos natively, an alternative way to get
conos running is through a docker container.
Note: On Mac OS X, Docker Machine has Memory and CPU limits. To control it, please check instructions either for CLI or for Docker Desktop.
The docker distribution has the latest version and also includes the
pagoda2 package.
To start a docker container, first install docker on your
platform and then start the pagoda2 container with the
following command in the shell:
docker run -p 8787:8787 -e PASSWORD=pass pkharchenkolab/conos:latestThe first time you run this command, it will download several large
images so make sure that you have fast internet access setup. You can
then point your browser to http://localhost:8787/ to get an Rstudio
environment with pagoda2 and conos installed
(please log in using credentials username=rstudio,
password=pass). Explore the docker –mount option
to allow access of the docker image to your local files.
Note: If you already downloaded the docker image and want to update it, please pull the latest image with:
docker pull pkharchenkolab/conos:latestIf you want to build image by your own, download the Dockerfile
(available in this repo under /docker) and run to following
command to build it:
docker build -t conos .This will create a “conos” docker image on your system (please be patient, as the build could take approximately 30-50 minutes to finish). You can then run it using the following command:
docker run -d -p 8787:8787 -e PASSWORD=pass --name conos -it conosIf you find this software useful for your research, please cite the corresponding paper:
Barkas N., Petukhov V., Nikolaeva D., Lozinsky Y., Demharter S., Khodosevich K., & Kharchenko P.V.
Joint analysis of heterogeneous single-cell RNA-seq dataset collections.
Nature Methods, (2019). doi:10.1038/s41592-019-0466-zThe R package can be cited as:
Viktor Petukhov, Nikolas Barkas, Peter Kharchenko, and Evan
Biederstedt (2021). conos: Clustering on Network of Samples. R
package version 1.4.7.